Publications
Scroll down for articles in descending order of publication date.
For a full list, please check Google Scholar.
Scroll down for articles in descending order of publication date.
For a full list, please check Google Scholar.
ChemRxiv (2025)
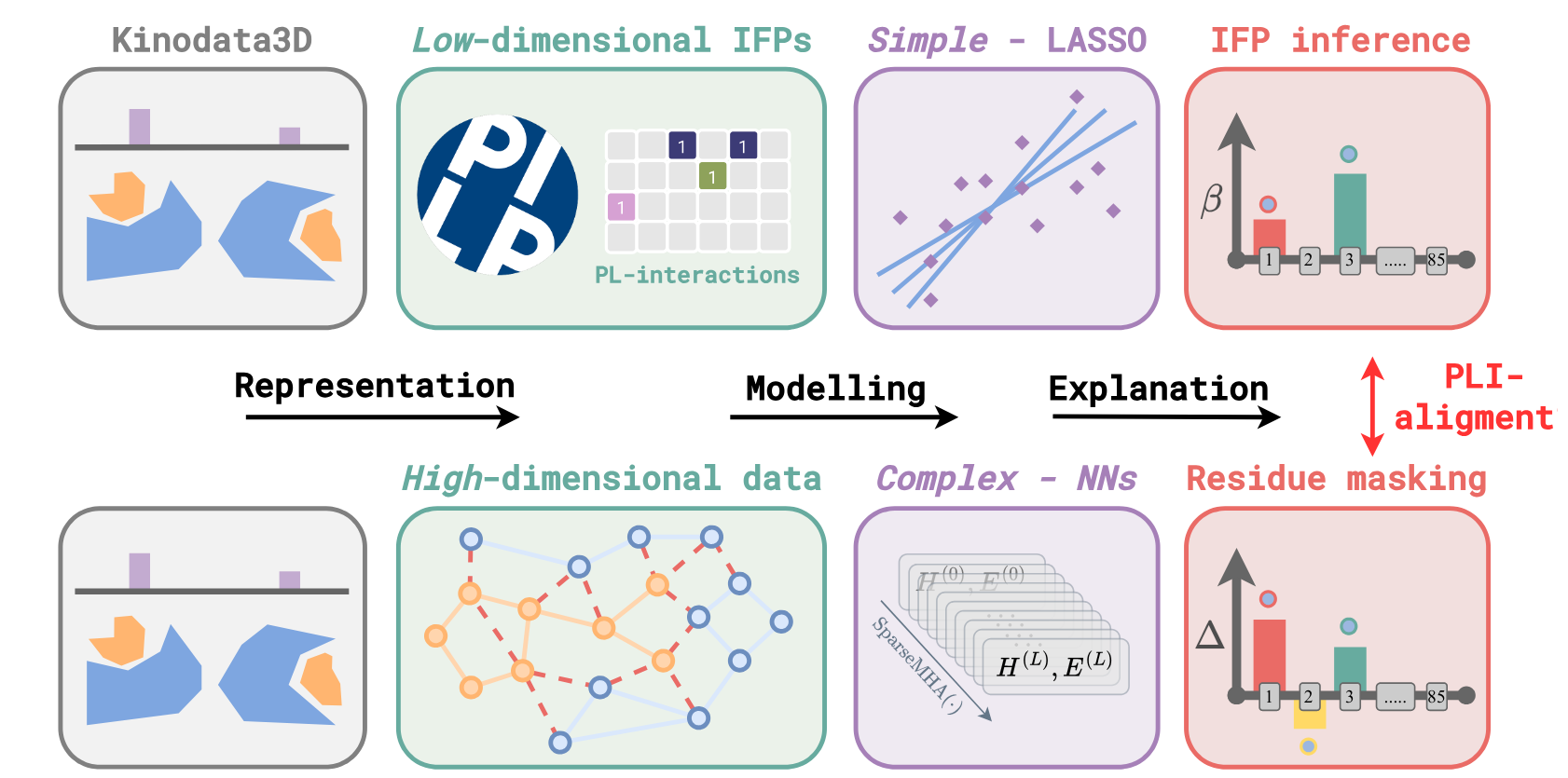
Accurately modeling interactions between small molecules and proteins using machine learning (ML) remains a central challenge in modern drug discovery. In particular, achieving and reliably assessing generalization in ML-based mod- els for binding affinity prediction has proven difficult. Focusing on the protein kinase domain, we investigate both the predictive performance and biophysical plausibility of graph neural networks (GNNs) trained to predict binding affinity, with and without access to docked protein–ligand complexes. To this end, we introduce a model-agnostic explainable AI framework that interprets model pre- dictions by attributing them to specific residues within the ATP-binding pocket of kinases. On the basis of discrete interaction fingerprints (IFPs), we employ robust statistical methods and feature selection to derive reference interaction profiles that serve as ground-truth explanations. Our analysis identifies 131 dis- tinct residues across 29 unique kinases that play key roles in linking IFP and binding affinity data. This enables a complementary benchmark, spanning nearly 10,000 docked complexes, to quantitatively assess model quality not only in terms of predictive accuracy but also biophysical alignment. The benchmark quantifies to what extent the model’s predictive mechanism aligns with meaningful bio- physical mechanisms—specifically, whether it recognizes protein regions actively involved in ligand binding. In a pilot study, we find that models incorporating 3D structural features exhibit moderate biophysical alignment, whereas baseline models without 3D information show no significant alignment, underscoring the structural nature of molecular binding, and importance thereof for training GNN models.
Living Journal of Computational Molecular Science (2025)
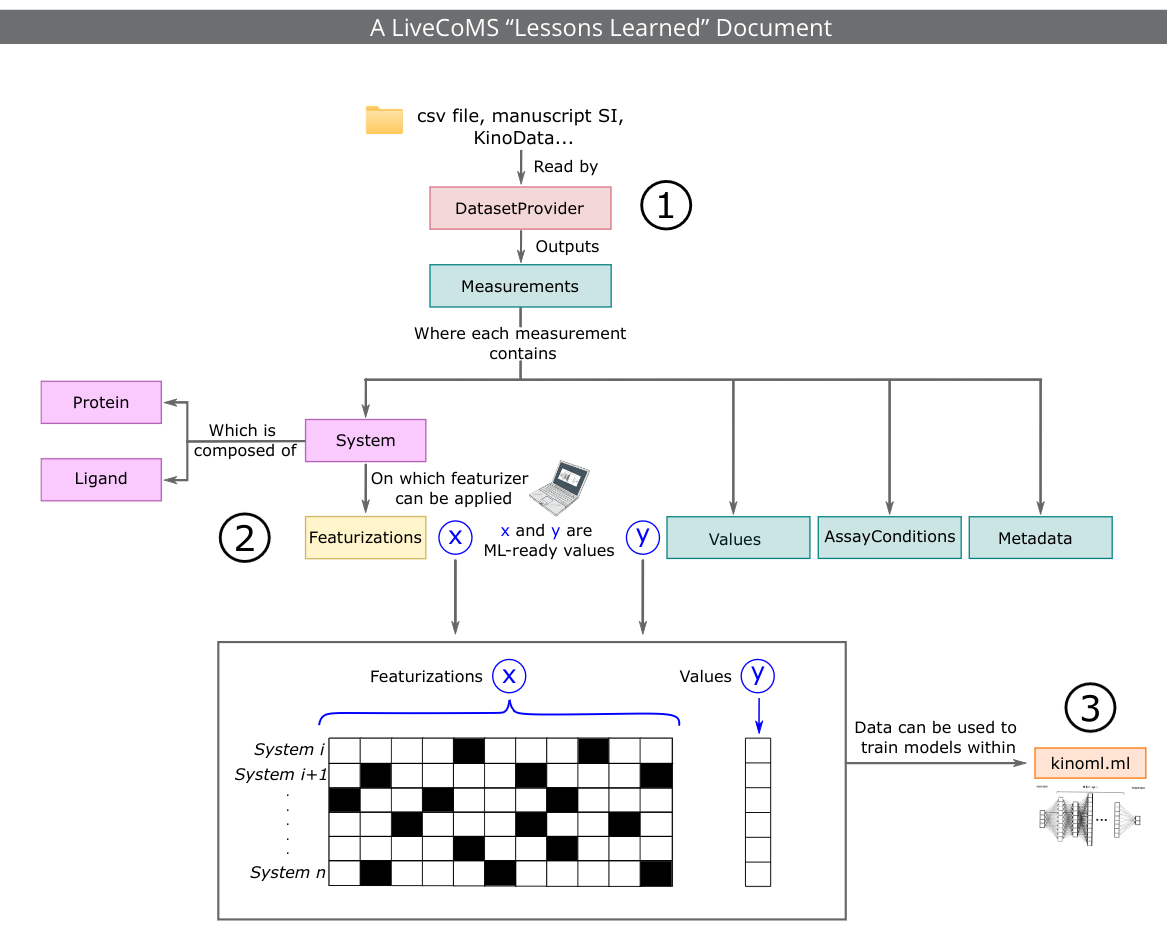
Recent advances in machine learning (ML) are reshaping drug discovery. Structure-based ML methods use physically-inspired models to predict binding affinities from protein:ligand complexes. These methods promise to enable the integration of data for many related targets, which addresses issues related to data scarcity for single targets and could enable generalizable predictions for a broad range of targets, including mutations. In this work, we report our experiences in building KinoML, a novel framework for ML in target-based small molecule drug discovery with an emphasis on structure-enabled methods. KinoML focuses currently on kinases as the relative structural conservation of this protein superfamily, particularly in the kinase domain, means it is possible to leverage data from the entire superfamily to make structure-informed predictions about binding affinities, selectivities, and drug resistance. Some key lessons learned in building KinoML include the importance of reproducible data collection and deposition, the harmonization of molecular data and featurization, and the selection of the right data format to ensure reusability and reproducibility of ML models. As a result, KinoML allows users to easily achieve three tasks: accessing and curating molecular data; featurizing this data with representations suitable for ML applications; and running reproducible ML experiments that require access to ligand, protein, and assay information to predict ligand affinity. Despite KinoML focusing on kinases, this framework can be applied to other proteins. The lessons reported here can help guide the development of platforms for structure-enabled ML in other areas of drug discovery.
PLOS Computational Biology (2025)
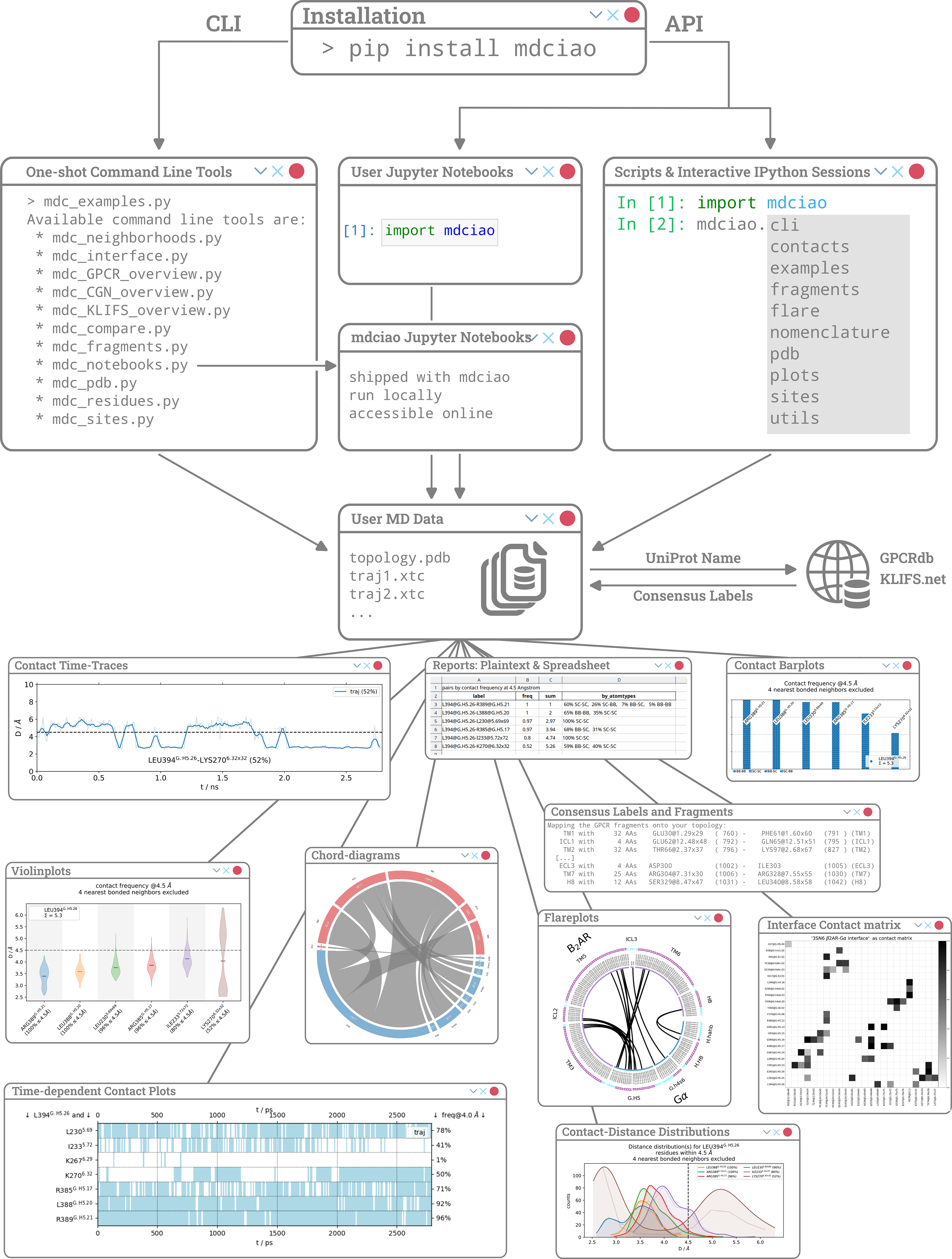
We present mdciao, an open-source command line tool and Python Application-Programming-Interface (API) for easy, one-shot analysis and representation of molecular dynamics (MD) simulation data. Building upon the widely used concept of residue-residue contact-frequencies, mdciao offers a wide spectrum of further analysis and representations, enriched with available domain specific annotations. The user-friendly interface offers pre-packaged solutions for non-expert users, while keeping customizability for expert ones. Emphasis has been put into automatically producing annotated, production-ready figures and tables. Furthermore, seamless on-the-fly query and inclusion of domain-specific generic residue numbering for GPCRs, GAIN-domains, G-proteins, and kinases is made possible through online lookups. This allows for easy selection and comparison across different systems, regardless of sequence identity, target residues or domains. Finally, the fully documented Python API allows users to include the basic or advanced mdciao functions in their analysis workflows, and provides numerous examples and Jupyter Notebook Tutorials. The source code is published under the GNU Lesser General Public License v3.0 or later and hosted on https://github.com/gph82/mdciao , and the documentation, including guides and examples, can be found at https://www.mdciao.org
Nature Communications (2025)
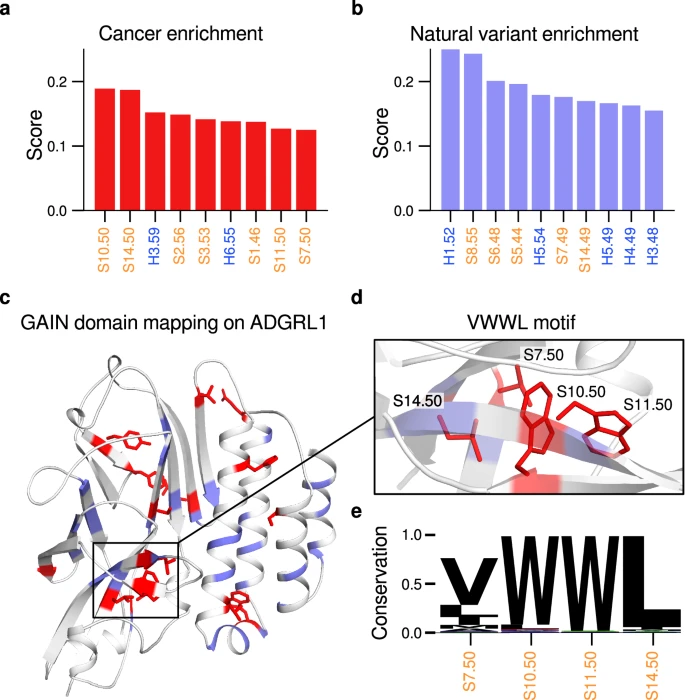
The GPCR autoproteolysis inducing (GAIN) domain is an ancient protein fold ubiquitous in adhesion G protein-coupled receptors (aGPCR). It contains a tethered agonist necessary and sufficient for receptor activation. The GAIN domain is a hotspot for pathological mutations. However, the low primary sequence conservation of GAIN domains has thus far hindered the knowledge transfer across different GAIN domains in human receptors as well as species orthologs. Here, we present a scheme for generic residue numbering of GAIN domains, based on structural alignments of over 14,000 modeled GAIN domain structures. This scheme is implemented in the GPCR database (GPCRdb) and elucidates the domain topology across different aGPCRs and their homologs in a large panel of species. We identify conservation hotspots and statistically cancer-enriched positions in human aGPCRs and show the transferability of positional and structural information between GAIN domain homologs. The GAIN-GRN scheme provides a robust strategy to allocate structural homologies at the primary and secondary levels also to GAIN domains of polycystic kidney disease 1/PKD1-like proteins, which now renders positions in both GAIN domain types comparable to one another. In summary, our work enables researchers to generate hypothesis and rationalize experiments related to GAIN domain function and pathology.
Nature Structural & Molecular Biology (2024)
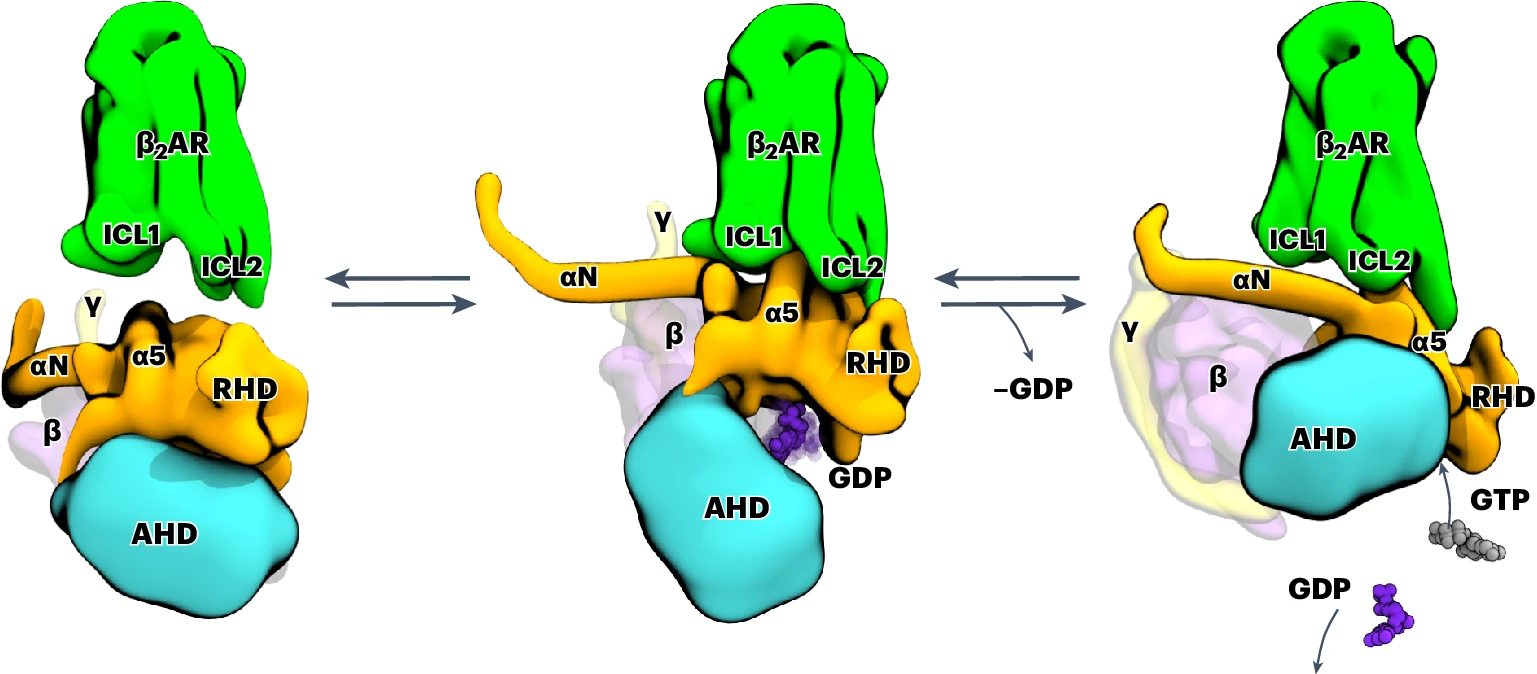
G-protein-coupled receptors (GPCRs) activate heterotrimeric G proteins by promoting guanine nucleotide exchange. Here, we investigate the coupling of G proteins with GPCRs and describe the events that ultimately lead to the ejection of GDP from its binding pocket in the Gα subunit, the rate-limiting step during G-protein activation. Using molecular dynamics simulations, we investigate the temporal progression of structural rearrangements of GDP-bound Gs protein (Gs·GDP; hereafter GsGDP) upon coupling to the β2-adrenergic receptor (β2AR) in atomic detail. The binding of GsGDP to the β2AR is followed by long-range allosteric effects that significantly reduce the energy needed for GDP release: the opening of α1-αF helices, the displacement of the αG helix and the opening of the α-helical domain. Signal propagation to the Gs occurs through an extended receptor interface, including a lysine-rich motif at the intracellular end of a kinked transmembrane helix 6, which was confirmed by site-directed mutagenesis and functional assays. From this β2AR–GsGDP intermediate, Gs undergoes an in-plane rotation along the receptor axis to approach the β2AR–Gsempty state. The simulations shed light on how the structural elements at the receptor–G-protein interface may interact to transmit the signal over 30 Å to the nucleotide-binding site. Our analysis extends the current limited view of nucleotide-free snapshots to include additional states and structural features responsible for signaling and G-protein coupling specificity.
Nature (2024)
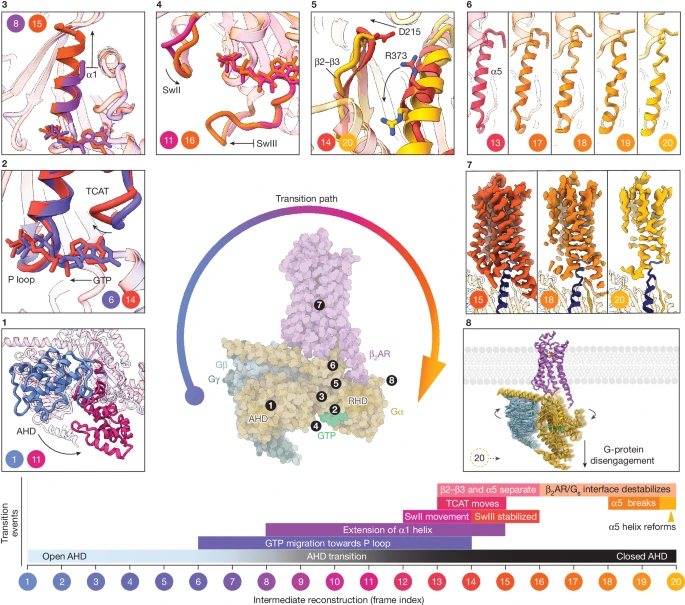
G-protein-coupled receptors (GPCRs) activate heterotrimeric G proteins by stimulating guanine nucleotide exchange in the Gα subunit1. To visualize this mechanism, we developed a time-resolved cryo-EM approach that examines the progression of ensembles of pre-steady-state intermediates of a GPCR–G-protein complex. By monitoring the transitions of the stimulatory Gs protein in complex with the β2-adrenergic receptor at short sequential time points after GTP addition, we identified the conformational trajectory underlying G-protein activation and functional dissociation from the receptor. Twenty structures generated from sequential overlapping particle subsets along this trajectory, compared to control structures, provide a high-resolution description of the order of main events driving G-protein activation in response to GTP binding. Structural changes propagate from the nucleotide-binding pocket and extend through the GTPase domain, enacting alterations to Gα switch regions and the α5 helix that weaken the G-protein–receptor interface. Molecular dynamics simulations with late structures in the cryo-EM trajectory support that enhanced ordering of GTP on closure of the α-helical domain against the nucleotide-bound Ras-homology domain correlates with α5 helix destabilization and eventual dissociation of the G protein from the GPCR. These findings also highlight the potential of time-resolved cryo-EM as a tool for mechanistic dissection of GPCR signalling events.
Nature Communications (2023)
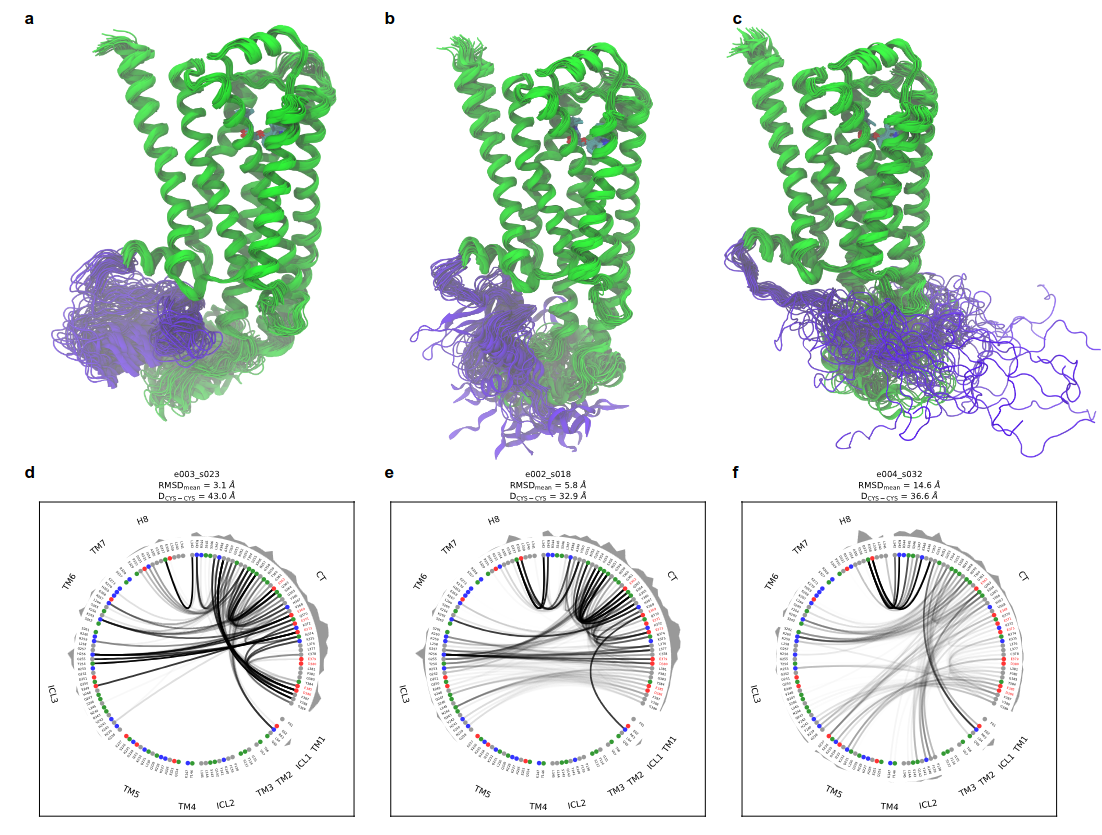
Advances in structural biology have provided important mechanistic insights into signaling by the transmembrane core of G-protein coupled receptors (GPCRs); however, much less is known about intrinsically disordered regions such as the carboxyl terminus (CT), which is highly flexible and not visible in GPCR structures. The β2adrenergic receptor’s (β2AR) 71 amino acid CT is a substrate for GPCR kinases and binds β-arrestins to regulate signaling. Here we show that the β2AR CT directly inhibits basal and agonist-stimulated signaling in cell lines lacking β-arrestins. Combining single-molecule fluorescence resonance energy transfer (FRET), NMR spectroscopy, and molecular dynamics simulations, we reveal that the negatively charged β2AR-CT serves as an autoinhibitory factor via interacting with the positively charged cytoplasmic surface of the receptor to limit access to G-proteins. The stability of this interaction is influenced by agonists and allosteric modulators, emphasizing that the CT plays important role in allosterically regulating GPCR activation.
Nucleic Acids Research (2022)
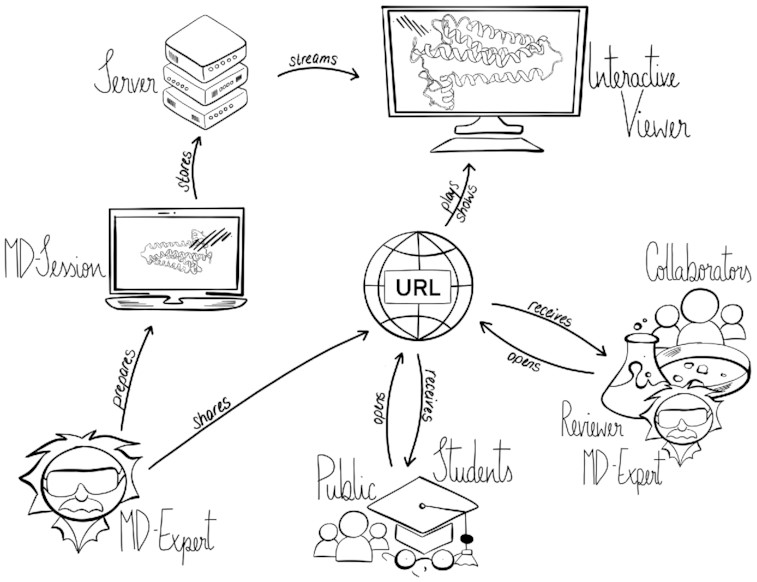
Molecular dynamics simulation is a proven technique for computing and visualizing the time-resolved motion of macromolecules at atomic resolution. The MDsrv is a tool that streams MD trajectories and displays them interactively in web browsers without requiring advanced skills, facilitating interactive exploration and collaborative visual analysis. We have now enhanced the MDsrv to further simplify the upload and sharing of MD trajectories and improve their online viewing and analysis. With the new instance, the MDsrv simplifies the creation of sessions, which allows the exchange of MD trajectories with preset representations and perspectives. An important innovation is that the MDsrv can now access and visualize trajectories from remote datasets, which greatly expands its applicability and use, as the data no longer needs to be accessible on a local server. In addition, initial analyses such as sequence or structure alignments, distance measurements, or RMSD calculations have been implemented, which optionally support visual analysis. Finally, based on Mol*, MDsrv now provides faster and more efficient visualization of even large trajectories compared to its predecessor tool NGL.
Molecular Cell (2021)
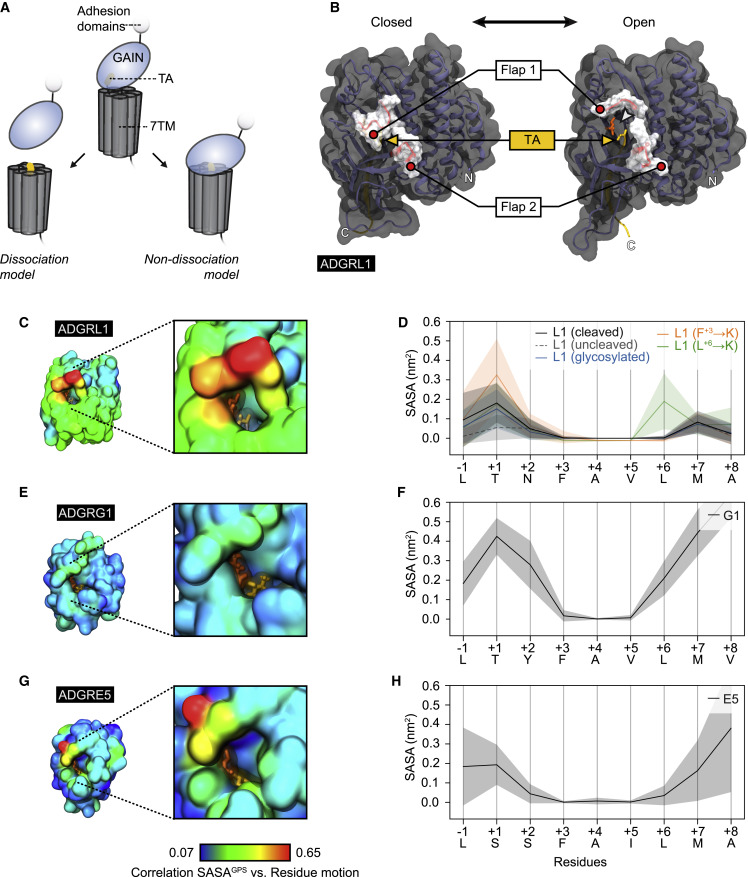
Adhesion G protein-coupled receptors (aGPCRs)/family B2 GPCRs execute critical tasks during development and the operation of organs, and their genetic lesions are associated with human disorders, including cancers. Exceptional structural aGPCR features are the presence of a tethered agonist (TA) concealed within a GPCR autoproteolysis-inducing (GAIN) domain and their non-covalent heteromeric two-subunit layout. How the TA is poised for activation while maintaining this delicate receptor architecture is central to conflicting signaling paradigms that either involve or exclude aGPCR heterodimer separation. We investigated this matter in five mammalian aGPCR homologs (ADGRB3, ADGRE2, ADGRE5, ADGRG1, and ADGRL1) and demonstrate that intact aGPCR heterodimers exist at the cell surface, that the core TA region becomes unmasked in the cleaved GAIN domain, and that intra-GAIN domain movements regulate the level of tethered agonist exposure, thereby likely controlling aGPCR activity. Collectively, these findings delineate a unifying mechanism for TA-dependent signaling of intact aGPCRs.
Scientific Reports (2018)
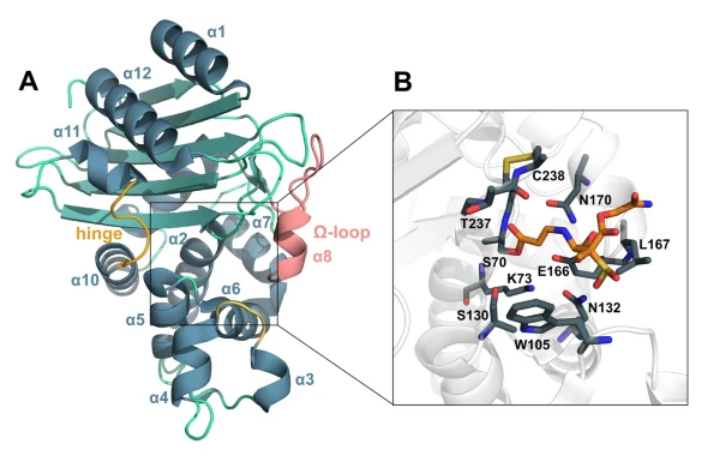
The rise of multi-drug resistance in bacterial pathogens is one of the grand challenges facing medical science. A major concern is the speed of development of β-lactamase-mediated resistance in Gram-negative species, thus putting at risk the efficacy of the most recently approved antibiotics and inhibitors, including carbapenems and avibactam, respectively. New strategies to overcome resistance are urgently required, which will ultimately be facilitated by a deeper understanding of the mechanisms that regulate the function of β-lactamases such as the Klebsiella Pneumoniae carbapenemases (KPCs). Using enhanced sampling computational methods together with site-directed mutagenesis, we report the identification of two “hydrophobic networks” in the KPC-2 enzyme, the integrity of which has been found to be essential for protein stability and corresponding resistance. Present throughout the structure, these networks are responsible for the structural integrity and allosteric signaling. Disruption of the networks leads to a loss of the KPC-2 mediated resistance phenotype, resulting in restored susceptibility to different classes of β-lactam antibiotics including carbapenems and cephalosporins. The ”hydrophobic networks” were found to be highly conserved among class-A β-lactamases, which implies their suitability for exploitation as a potential target for therapeutic intervention.
Journal of chemical theory and computation (2017)
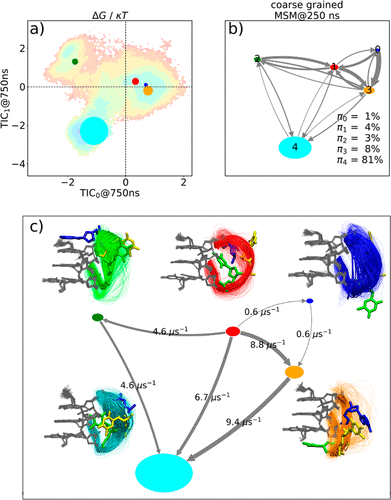
We have carried out a series of extended unbiased molecular dynamics (MD) simulations (up to 10 μs long, ∼162 μs in total) complemented by replica-exchange with the collective variable tempering (RECT) approach for several human telomeric DNA G-quadruplex (GQ) topologies with TTA propeller loops. We used different AMBER DNA force-field variants and also processed simulations by Markov State Model (MSM) analysis. The slow conformational transitions in the propeller loops took place on a scale of a few μs, emphasizing the need for long simulations in studies of GQ dynamics. The propeller loops sampled similar ensembles for all GQ topologies and for all force-field dihedral-potential variants. The outcomes of standard and RECT simulations were consistent and captured similar spectrum of loop conformations. However, the most common crystallographic loop conformation was very unstable with all force-field versions. Although the loss of canonical γ-trans state of the first propeller loop nucleotide could be related to the indispensable bsc0 α/γ dihedral potential, even supporting this particular dihedral by a bias was insufficient to populate the experimentally dominant loop conformation. In conclusion, while our simulations were capable of providing a reasonable albeit not converged sampling of the TTA propeller loop conformational space, the force-field description still remained far from satisfactory.
Journal of Chemical Theory and Computation (2016)
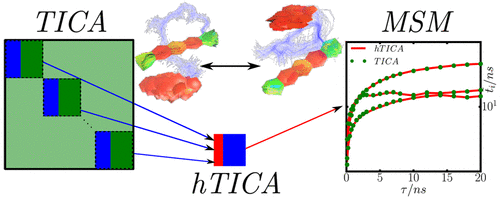
Analysis of molecular dynamics, for example using Markov models, often requires the identification of order parameters that are good indicators of the rare events, i.e. good reaction coordinates. Recently, it has been shown that the time-lagged independent component analysis (TICA) finds the linear combinations of input coordinates that optimally represent the slow kinetic modes and may serve in order to define reaction coordinates between the metastable states of the molecular system. A limitation of the method is that both computing time and memory requirements scale with the square of the number of input features. For large protein systems, this exacerbates the use of extensive feature sets such as the distances between all pairs of residues or even heavy atoms. Here we derive a hierarchical TICA (hTICA) method that approximates the full TICA solution by a hierarchical, divide-and-conquer calculation. By using hTICA on distances between heavy atoms we identify previously unknown relaxation processes in the bovine pancreatic trypsin inhibitor.
Journal of Chemical Theory and Computation (2015)
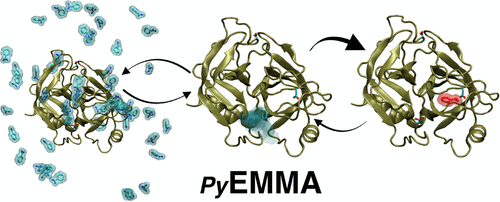
Markov (state) models (MSMs) and related models of molecular kinetics have recently received a surge of interest as they can systematically reconcile simulation data from either a few long or many short simulations and allow us to analyze the essential metastable structures, thermodynamics, and kinetics of the molecular system under investigation. However, the estimation, validation, and analysis of such models is far from trivial and involves sophisticated and often numerically sensitive methods. In this work we present the open-source Python package PyEMMA (http://pyemma.org) that provides accurate and efficient algorithms for kinetic model construction. PyEMMA can read all common molecular dynamics data formats, helps in the selection of input features, provides easy access to dimension reduction algorithms such as principal component analysis (PCA) and time-lagged independent component analysis (TICA) and clustering algorithms such as k-means, and contains estimators for MSMs, hidden Markov models, and several other models. Systematic model validation and error calculation methods are provided. PyEMMA offers a wealth of analysis functions such that the user can conveniently compute molecular observables of interest. We have derived a systematic and accurate way to coarse-grain MSMs to few states and to illustrate the structures of the metastable states of the system. Plotting functions to produce a manuscript-ready presentation of the results are available. In this work, we demonstrate the features of the software and show new methodological concepts and results produced by PyEMMA.
Journal of Chemical Theory and Computation (2014)
The eigenvalues and eigenvectors of the molecular dynamics propagator (or transfer operator) contain the essential information about the molecular thermodynamics and kinetics. This includes the stationary distribution, the metastable states, and state-to-state transition rates. Here, we present a variational approach for computing these dominant eigenvalues and eigenvectors. This approach is analogous to the variational approach used for computing stationary states in quantum mechanics. A corresponding method of linear variation is formulated. It is shown that the matrices needed for the linear variation method are correlation matrices that can be estimated from simple MD simulations for a given basis set. The method proposed here is thus to first define a basis set able to capture the relevant conformational transitions, then compute the respective correlation matrices, and then to compute their dominant eigenvalues and eigenvectors, thus obtaining the key ingredients of the slow kinetics.
The Journal of Chemical Physics (2013)
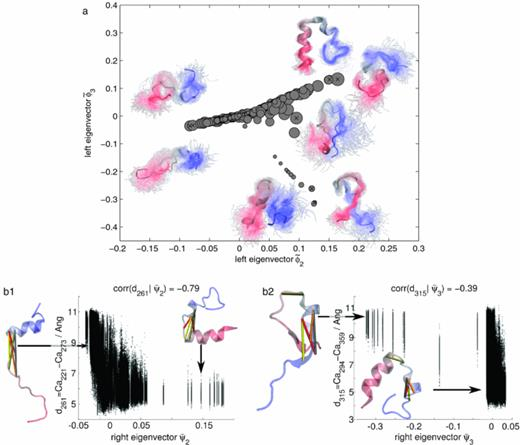
A goal in the kinetic characterization of a macromolecular system is the description of its slow relaxation processes via (i) identification of the structural changes involved in these processes and (ii) estimation of the rates or timescales at which these slow processes occur. Most of the approaches to this task, including Markov models, master-equation models, and kinetic network models, start by discretizing the high-dimensional state space and then characterize relaxation processes in terms of the eigenvectors and eigenvalues of a discrete transition matrix. The practical success of such an approach depends very much on the ability to finely discretize the slow order parameters. How can this task be achieved in a high-dimensional configuration space without relying on subjective guesses of the slow order parameters? In this paper, we use the variational principle of conformation dynamics to derive an optimal way of identifying the “slow subspace” of a large set of prior order parameters – either generic internal coordinates or a user-defined set of parameters. Using a variational formulation of conformational dynamics, it is shown that an existing method—the time-lagged independent component analysis—provides the optional solution to this problem. In addition, optimal indicators—order parameters indicating the progress of the slow transitions and thus may serve as reaction coordinates—are readily identified. We demonstrate that the slow subspace is well suited to construct accurate kinetic models of two sets of molecular dynamics simulations, the 6-residue fluorescent peptide MR121-GSGSW and the 30-residue intrinsically disordered peptide kinase inducible domain (KID). The identified optimal indicators reveal the structural changes associated with the slow processes of the molecular system under analysis.
Physical Chemistry Chemical Physics (2013)
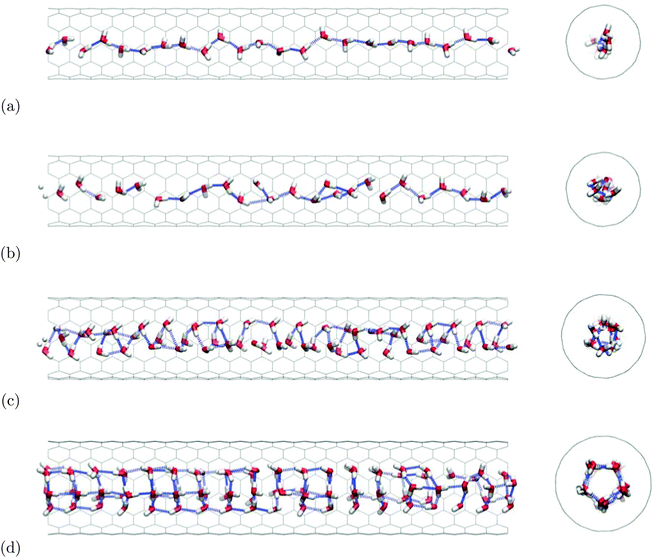
Effective Lennard-Jones models for the water–carbon interaction are derived from existing high-level ab initio calculations of water adsorbed on graphene models. The resulting potential energy well (εCO + 2εCH ≈ 1 kJ mol−1) is deeper than most of the previously used values in the literature on water in carbon nanotubes (CNTs). Moreover, a substantial anisotropy of the water–carbon interaction (εCO ≈ 2εCH) is obtained, which is neglected in most of the literature. We systematically investigate the effect of this anisotropy on structure and dynamics of TIP5P water confined in narrow, single-walled CNTs by means of molecular dynamics simulations for T = 300 K. While for isotropic models water usually forms one-dimensional, ordered chains inside (6,6) CNTs, we find frequent chain ruptures in simulations with medium to strongly anisotropic potentials. Here, the water molecules tend to form denser clusters displaying a liquid-like behaviour, allowing for self-diffusion along the CNT axis, in contrast to all previous simulations employing spherical (εCH = 0) interaction models. For (7,7) CNTs we observe structures close to trigonal, helical ice nanotubes which exhibit a non-monotonous dependence on the anisotropy of the water–carbon interaction. Both for vanishing and for large values of εCH we find increased fluctuations leading to a more liquid-like behaviour, with enhanced axial diffusion. In contrast, structure and dynamics of water inside (8,8) CNTs are found to be almost independent of the anisotropy of the underlying potential, which is attributed to the higher stability of the non-helical fivefold water prisms. We predict this situation to also prevail for larger CNTs, as the influence of the water–water interaction dominates over that of the water–carbon interaction.
Theoretical Chemistry Accounts (2012)
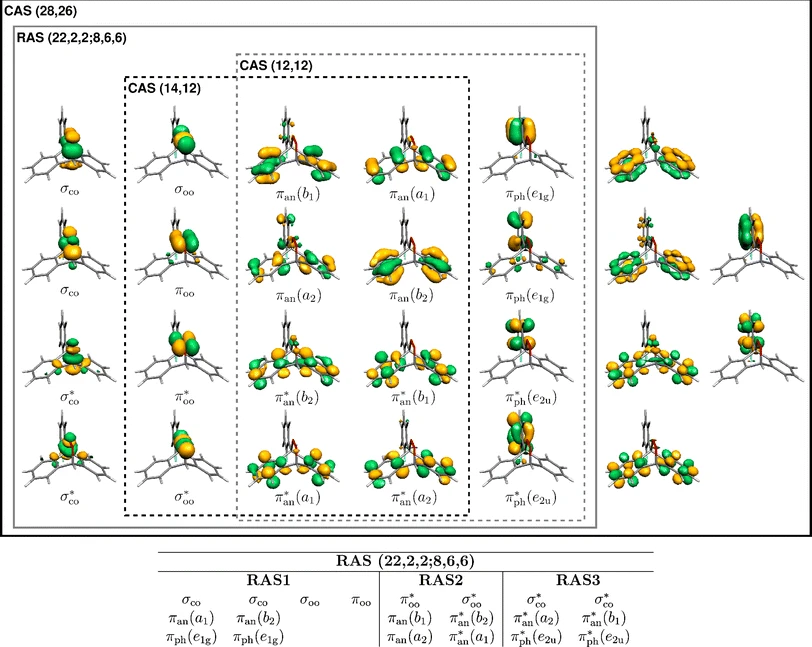
The different electronic excited states of o-fluorine-phenyl-9-anthracene-9,10-endoperoxide have been benchmarked with RASPT2. The lowest excited state corresponds to the homolytic O–O dissociation and higher excited states are connected to singlet oxygen generation.
The Journal of Physical Chemistry A (2012)
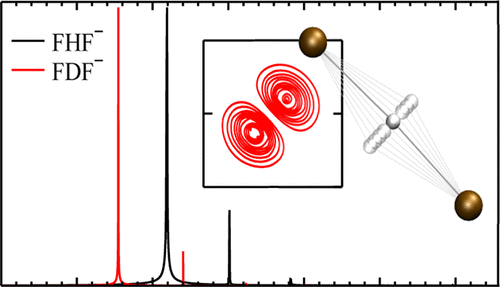
A four dimensional (4D) time-dependent calculation to obtain the first vibrational states of the hydrogen bifluoride ion, FHF–, and its deuterated counterpart, FDF–, has been performed using a spectral method in Cartesian coordinates. The corresponding potential energy surfaces have been computed at the CCSD(T)/aug-cc-pVTZ level of theory. The obtained values for the fundamental vibrational bands ν̃1 = 589 cm–1, ν̃2 = 1305 cm–1, and ν̃3 = 1372 cm–1 assigned to the symmetric stretch, bend, and asymmetric stretch modes, respectively (598, 943, and 972 cm–1 for FDF–, respectively) are in good agreement with available experimental and theoretical values. Selected overtones and mixed modes are also calculated. Infrared spectra have been simulated using the dipole approximation for two different polarization directions of the incident light.
Chemical Physics (2010)
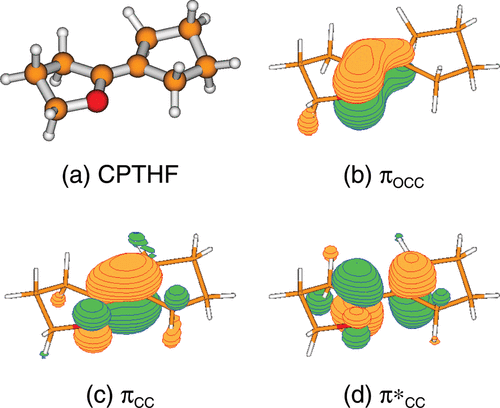
The excited state dynamics of 2-cyclopentylidene-tetrahydrofuran (CPTHF) is investigated using quantum dynamics. CPTHF can be considered a model for an asymmetric molecular rotor in which unidirectional rotation could be triggered around the double bond. After excitation, conical intersections at twisted angles allow for rationless decay to the ground state. Two-dimensional potential energy surfaces for the ground and first (ππ∗) excited state have been calculated using CASSCF. They include the torsion around the double bond and the pyramidalisation at one carbon atom. The relaxation of CPTHF after photo-excitation has been then studied using up to five degrees of freedom. 2D wavepacket propagations on the explicit PESs do not allow the dissipation of the energy of the system after excitation. The inclusion of further modes, studied using the MCTDH method, show that the internal conversion rate is significantly altered depending on the modes included.
The Journal of Physical Chemistry A (2010)
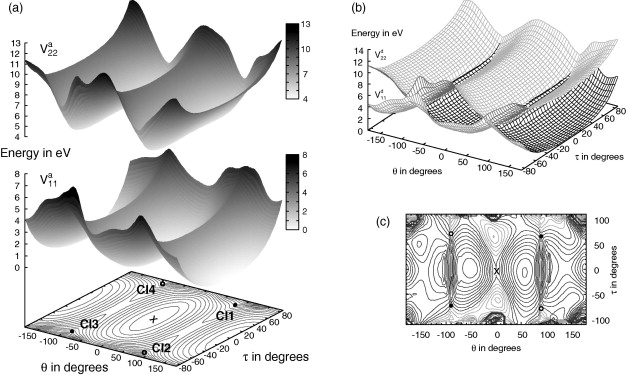
The ground state potential energy surface of the model molecular rotor 2-cyclopentylidene-tetrahydrofuran (CPTHF) has been characterized by calculating minimum energy conformations, racemization pathways, and rotational barriers with high level ab initio electronic structure calculations. Two conformers with their corresponding enantiomers are found. The activation barriers for racemization are negligible, therefore thermal racemization takes place at room temperature. Torsional transition states, calculated using multiconfigurational CASSCF calculations, show twisted and pyramidalized biradical structures. Additionally, the photochemistry of CPTHF has been investigated using the accurate MS-CASPT2/CASSCF methodology. In the UV spectrum it is found that the spectroscopic state is the S1, which corresponds to a ππ* transition within the ethylene moiety. To understand light-triggered isomerization around the C═C bond, five conical intersections between the S0 and S1 have been located for each conformer of CPTHF, which allow the system to rapidly decay to the electronic ground state.