Publications
Scroll down for articles in descending order of publication date.
For a full list, please check Google Scholar.
Scroll down for articles in descending order of publication date.
For a full list, please check Google Scholar.
Chemical Physics (2010)
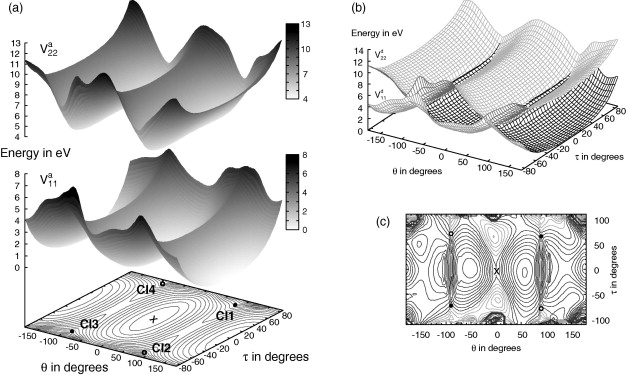
The excited state dynamics of 2-cyclopentylidene-tetrahydrofuran (CPTHF) is investigated using quantum dynamics. CPTHF can be considered a model for an asymmetric molecular rotor in which unidirectional rotation could be triggered around the double bond. After excitation, conical intersections at twisted angles allow for rationless decay to the ground state. Two-dimensional potential energy surfaces for the ground and first (ππ∗) excited state have been calculated using CASSCF. They include the torsion around the double bond and the pyramidalisation at one carbon atom. The relaxation of CPTHF after photo-excitation has been then studied using up to five degrees of freedom. 2D wavepacket propagations on the explicit PESs do not allow the dissipation of the energy of the system after excitation. The inclusion of further modes, studied using the MCTDH method, show that the internal conversion rate is significantly altered depending on the modes included.
The Journal of Physical Chemistry A (2010)
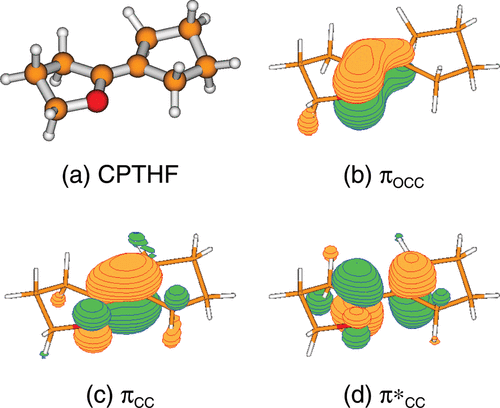
The ground state potential energy surface of the model molecular rotor 2-cyclopentylidene-tetrahydrofuran (CPTHF) has been characterized by calculating minimum energy conformations, racemization pathways, and rotational barriers with high level ab initio electronic structure calculations. Two conformers with their corresponding enantiomers are found. The activation barriers for racemization are negligible, therefore thermal racemization takes place at room temperature. Torsional transition states, calculated using multiconfigurational CASSCF calculations, show twisted and pyramidalized biradical structures. Additionally, the photochemistry of CPTHF has been investigated using the accurate MS-CASPT2/CASSCF methodology. In the UV spectrum it is found that the spectroscopic state is the S1, which corresponds to a ππ* transition within the ethylene moiety. To understand light-triggered isomerization around the C═C bond, five conical intersections between the S0 and S1 have been located for each conformer of CPTHF, which allow the system to rapidly decay to the electronic ground state.
New Journal of Physics (2010)
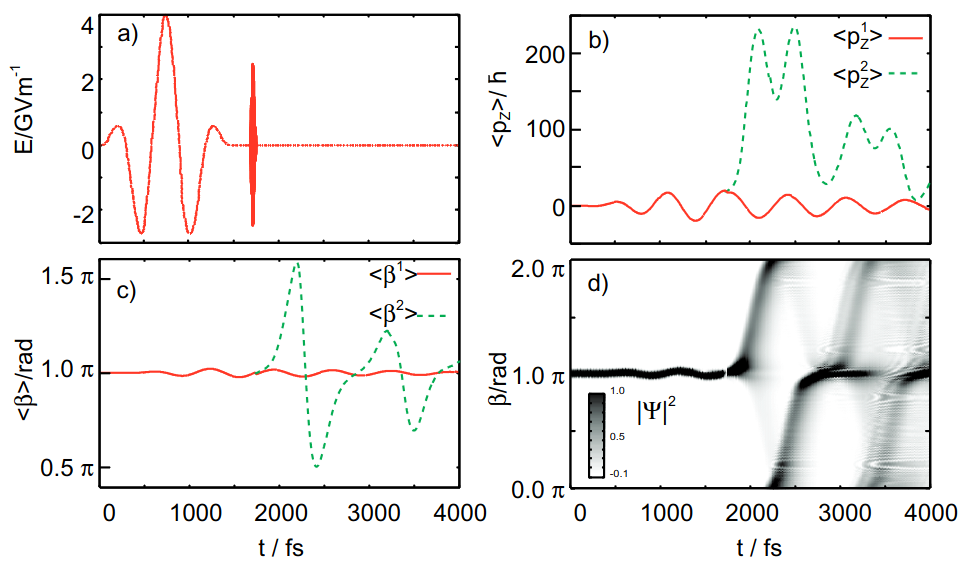
We investigate the extent to which unidirectional intramolecular torsional motion can be created in an oriented bicyclic model system driven solely by laser light. We apply the machinery of quantum control via specifically tailored laser pulses to induce such motion, eliminating the need for the thermally constrained steps conventionally used in molecular motor systems. Our approach does not rely on specific details of the potential surfaces to create a preferred direction. Rather, we use matter–field interaction and the tools of coherent optimal control to create a wave packet with nonzero angular momentum among unbound torsional states on an excited electronic surface. Analysis of the results of the control algorithm provides general insight into when and how optimal control theory can find solutions that could not be generated through simple intuitive schemes. We find that, under constrained polarization, the control algorithm reduces to a simple intuitive coherent control strategy wherein a first IR pulse creates a non-stationary wave packet on the ground surface and a subsequent UV pulse transfers it to the excited state. Allowing for polarization shaping, however, we find new control routes that go beyond the intuitive scheme.
Physical Chemistry Chemical Physics (2010)
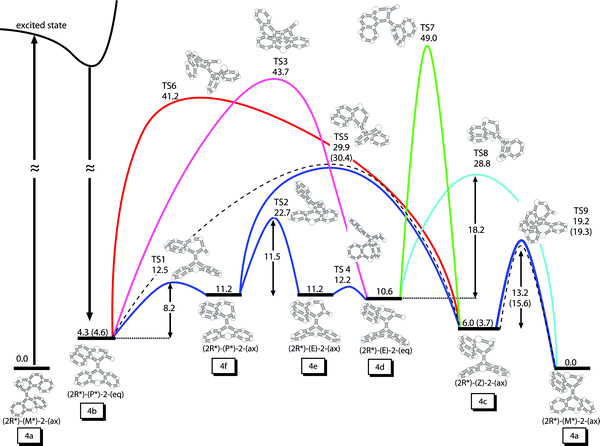
Chiral overcrowded alkenes are capable of unidirectional rotation via a series of cis-trans photochemical and helix-inversion thermal steps. Using a pseudo-random conformational search we have located different ground state minima belonging to the potential energy surface of two different overcrowded alkenes that function as molecular rotors. The transition states connecting the minima allow identifying different reaction pathways which are possible in the thermal helix-inversion steps. The mechanisms found for the two studied molecular rotors are different and provide a valuable insight into the conformational dynamics of the rotary cycle. While in one case the thermal step occurs via a single transition state, in the other, several intermediates are accessible. The associated energy barriers are in agreement with the experimental values, supporting the proposed mechanisms.
ChemPhysChem (2008)
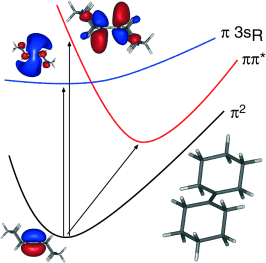
The electronic excited states of the olefin 1,1′‐bicylohexylidene (BCH) are investigated using multiconfigurational complete active space self‐consistent‐field second order perturbation theory in its multi‐state version (MS‐CASPT2). Our calculations undoubtedly show that the bulk of the intensity of the two unusually intense bands of the UV absorption of BCH measured with maxima at 5.95 eV and 6.82 eV in the vapor phase are due to a single ππ* valence excitation. Sharp peaks reported in the vicinity of the low‐energy feature in the gas phase correspond to the beginning of the π3sR Rydberg series. By locating the origin of the ππ* band at 5.63 eV, the intensity and broadening of the observed bands and their presence in solid phase is explained as the vibrational structure of the valence ππ* transition, which underlies the Rydberg manifold as a quasi‐continuum.
The Journal of Physical Chemistry B (2008)
The UV−vis absorption spectra of the photoreceptor chromophores biliverdin (BV) in the ZZZssa conformation and the phycocyanobilin (PCB) with conformations ZZZssa and ZZZasa have been investigated by means of time-dependent density functional theory (TD-DFT) with a polarized continuum model. The three systems are studied in different conditions to include protonation, solvation- and protein-environmental effects on gas phase and available X-ray structures. The crystal structures of BV in bacteriophytochrome of Deinococcus radiodurans and PCB in C-Phycocyanin serve to calibrate the performance of the TD-DFT method and allow estimating the spectral shifts created when gas phase structures instead of a proper environment are used. In contrast, the structure of PCB in the cyanobacterial phytochrome Cph1 is unknown. The excellent agreement of the theoretical spectrum with experimentally recorded data for the PCB in the cyanobacterial phytochrome Cph1 strongly supports a semicyclic ZZZssa structure, similar to that found for the BV chromophore.
Zeitschrift für Physikalische Chemie (2008)
We present hydrogen bonds with toroidal densities of the protons, called toroidal hydrogen bonds, in systems with cylindrical symmetry e.g. triatomic molecules AHB or ions AHB+ or AHB–. These may be prepared by excitation of the degenerate bending vibrations and related pseudorotation such that the hydrogen atom (or the corresponding proton) is no longer located on the symmetry axis between atoms A and B, but – classically speaking – it rotates around that axis. Quantum mechanically, the toroidal hydrogen bond is represented by an excited nuclear eigenstate with nuclear wavefunction and corresponding nuclear density which have toroidal shapes around the central nodal line which conincides with the AB symmetry axis. The properties of these bonds are analyzed, including the pseudorotational angular momentum. Toroidal hydrogen bonds may be excited by means of circularly polarized infrared (IR) laser pulses. The results are demonstrated exemplarily for the oriented model system FHF–, by means of combined quantum chemistry calculations of the potential energy and dipole surfaces (adapted from L. González, G. Pérez-Hernández, J. González-Vázquez, (2008), submitted), calculations of the vibrational and pseudorotational states in the frame of Watson’s isomorphic Hamiltonian for linear molecules (J. K. G. Watson, Mol. Phys. 19 (1970) 465), and quantum dynamics simulations of the laser driven nuclear dynamics, analogous to recent applications to CdH2 (I. Barth, J. Manz, P. Sebald, Chem. Phys. 346 (2008) 89).
AIP Conference Proceedings (2007)
Control of time‐dependent wavepackets in the frame of unimolecular reactions is described. The shown applications include photodissociation and photoisomerization dynamics in three isolated systems.