Scientific Software Contributions
Research Software In Context
PyEMMA
![]()
After completing my PhD in Quantum Chemistry and Quantum Reaction Dynamics (writing mostly FORTRAN90), in my early postdoc I had the opportunity to contribute to the release of PyEMMA 2 to the community, an effort led by Prof. Frank Noé.
After years of coding, this was my actual first exposure to scientific software best practices, which was eye-opening in matters like CI/CD and full development cycles. In 2015, we released the PyEMMA package and manuscript.
Working on PyEMMA also meant engaging a lot with the community via workshops and seminars, with the added benefit of allowing me to help other researchers analyze their own data, something that thoroughly enjoy until today.
I contributed mostly to the featurization aspects of MSM building, focusing on input metrics and their subsequent transformation that could yield good (or good enough) descriptors of conformational dynamics, which is a focus of my research.
Typically, these descriptors would get further transformed (e.g. with TICA, which I also contributed to) prior to MSM construction, but sooner or later, the insights gained from model-building always needed to be mapped back to the world of meaningful coordinates, something far from trivial in many cases. In that spirit, I developed a package, molPX, to visually explore what these projections meant.
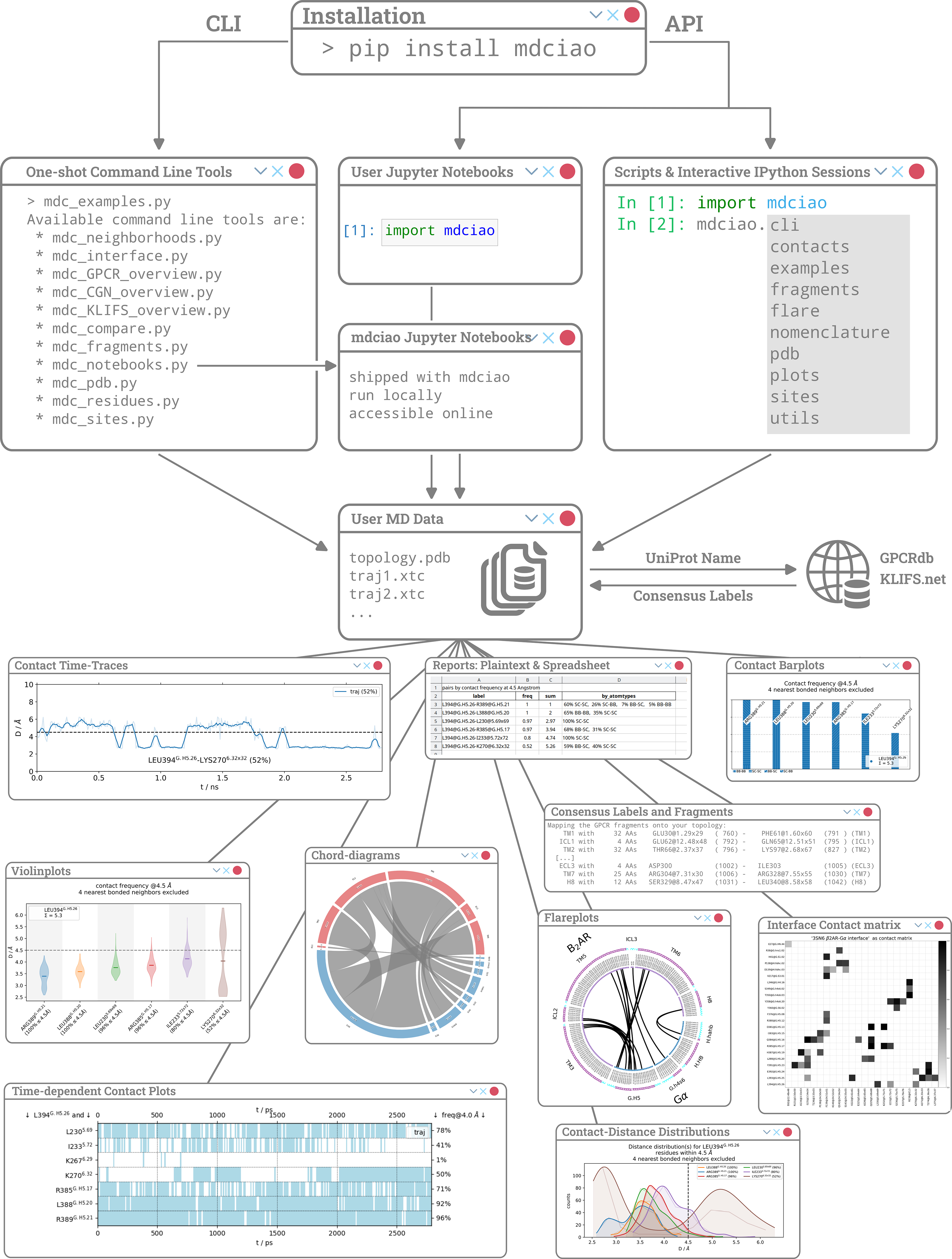
mdciao
![]()
When I arrived at Charité Universitätsmedizin Berlin, to the group of Prof. Peter W. Hildebrand (now in Leipzig), I started to work closely with more applied MD practitioners or even physicians-turned-structural-biologists like Prof. Brian Kobilka.
Here, the approach to MD analysis was focused more on summary statistics and differences in the behaviour of individual residues or protein regions. These regions were targeted for analysis because, among other things, they were directly accessible for experimental manipulation -by our collaborators themselves- or because of specific domain knowledge, like motifs associated with pathological mutations or signalling pathways.
With this in mind, I started to develop a collection of Python scripts to empower this kind of MD user, while also beginning my own journey of incorporating domain knowledge (about GPCRs, G-proteins, Kinases) into MD analysis software, to make it usable (and transferable across systems!) by non-expert users.
When the COVID pandemic hit, maintaining predictable working hours became a challenge, particularly for those of us with family obligations, making coordinated team efforts harder to sustain. So, I focused my working hours to shaping this collection of Python scripts into a modest, but proper, Python package: mdciao. It had its first PyPi release in November 2021 and BioRxiv preprint in July 2022. It has been nicely picking up citations since then, and it is always fun seeing it out in the wild.
When life went back to normal and projects started to pick up speed again, most of my time went back to interpreting -and helping others interpret- MD data, incorporating anything I learned in those projects into mdciao. After getting these publications out (with the help of mdciao), I finally managed to get the package its peer-reviewed publication in PLoS Comp Bio in 2025.
-
Function and dynamics of the intrinsically disordered carboxyl terminus of β2 adrenergic receptor
Heng, J., Hu, Y., Pérez-Hernández, G., et al.
Nature Communications 14, 2005 (2023) -
Time-resolved cryo-EM of G-protein activation by a GPCR
Papasergi-Scott, M.M., Pérez-Hernández, G., Batebi, H., et al.
Nature 629, 1182–1191 (2024) -
Mechanistic insights into G-protein coupling with an agonist-bound G-protein-coupled receptor
Pérez-Hernández, G., Batebi, H., Rahman, S.N., et al.
Nature Structural & Molecular Biology (2024) -
Generic residue numbering of the GAIN domain of adhesion GPCRs
Seufert, F., Pérez-Hernández, G., Pándy-Szekeres, G., et al.
Nature Communications 16, 246 (2025)