Graphical abstract: Highlights • Modular pipeline builder enabling fully automated, FAIR-compliant analyses. • User-friendly API that executes complete workflows with minimal code. • Efficient handling of large trajectories without requiring high-performance hardware. • End-to-end framework for featurization, decomposition, clustering and visualization. • Explainability using ML-based feature selection for key structural descriptors.
DOI
10.1016/j.jmb.2026.169809
Abstract
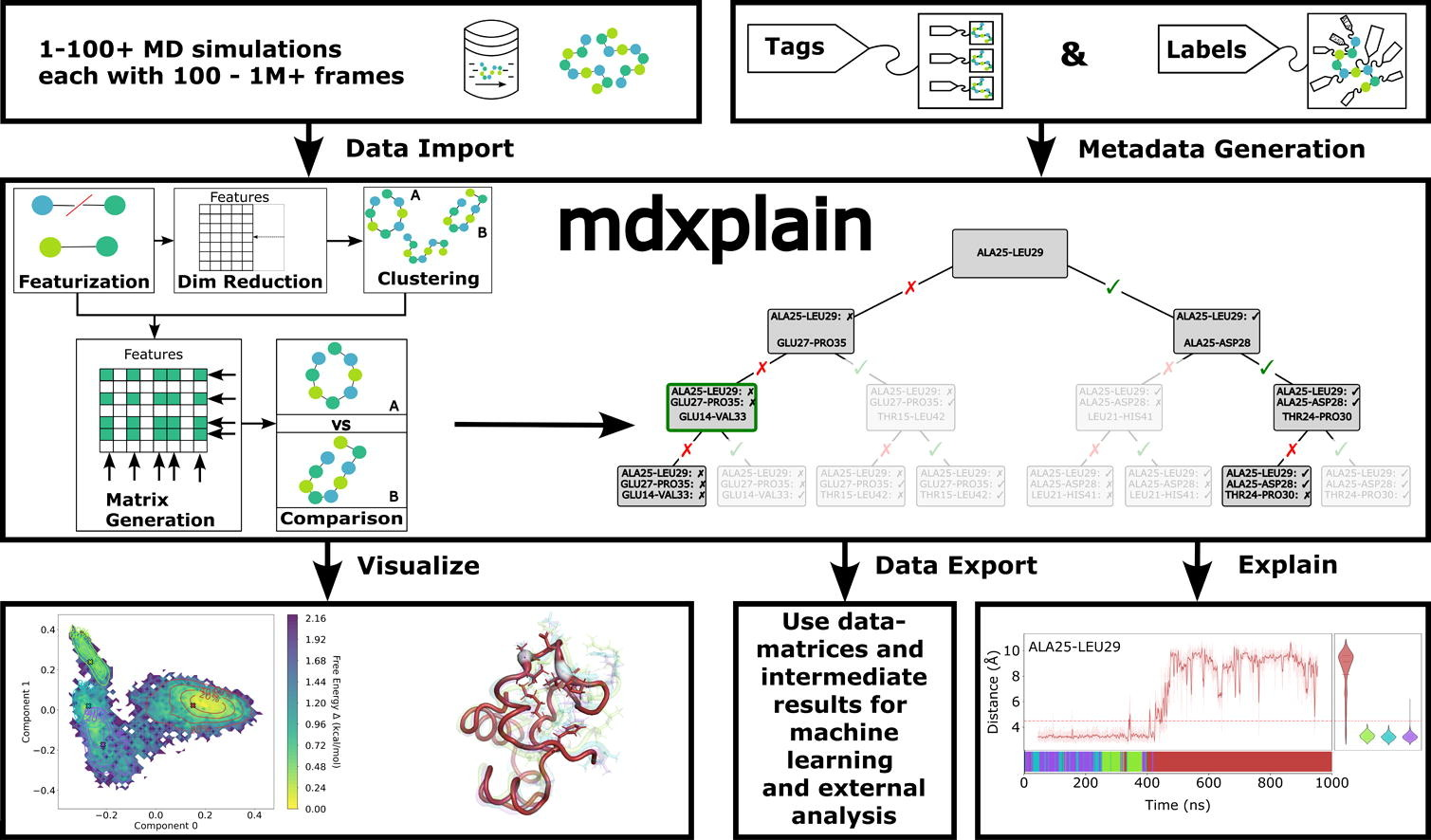
Molecular dynamics (MD) simulations provide detailed, time-resolved insight into molecular motion. Advances in hardware and software now make very large systems accessible, increasing the need for efficient tools to analyze the resulting trajectories. We introduce mdxplain, a high-level Python API that facilitates the creation of scalable, streamlined, and reusable analysis pipelines for large MD datasets with only a few lines of code. A unified object exposes all functionality, combining typical MD featurization and MD metrics with dimensionality reduction, clustering and feature selection via decision trees, supporting expert and non-expert users in identifying structural patterns and explain the dynamic behavior of their systems. Leveraging metadata annotations for trajectory- and residue-selection, mdxplain can handle multiple topologies in a single execution and uses optimized memory handling to process large datasets (millions of frames) efficiently. Its reports include distributional and time-series plots, representative conformations and decision trees combined with optional 3D visualization via PyMOL and NGLView. Pipelines can be exported at all time, bundling all relevant data for sharing and reuse, ensuring reproducibility and FAIR compliance. The Python API, together with documentation, examples, and tutorials is available on github.com/maximilian-salomon/mdxplain and on mdxplain.de.