Fig. 1: Graphical abstract
DOI
10.26434/chemrxiv-2025-qsw7v-v3
Abstract
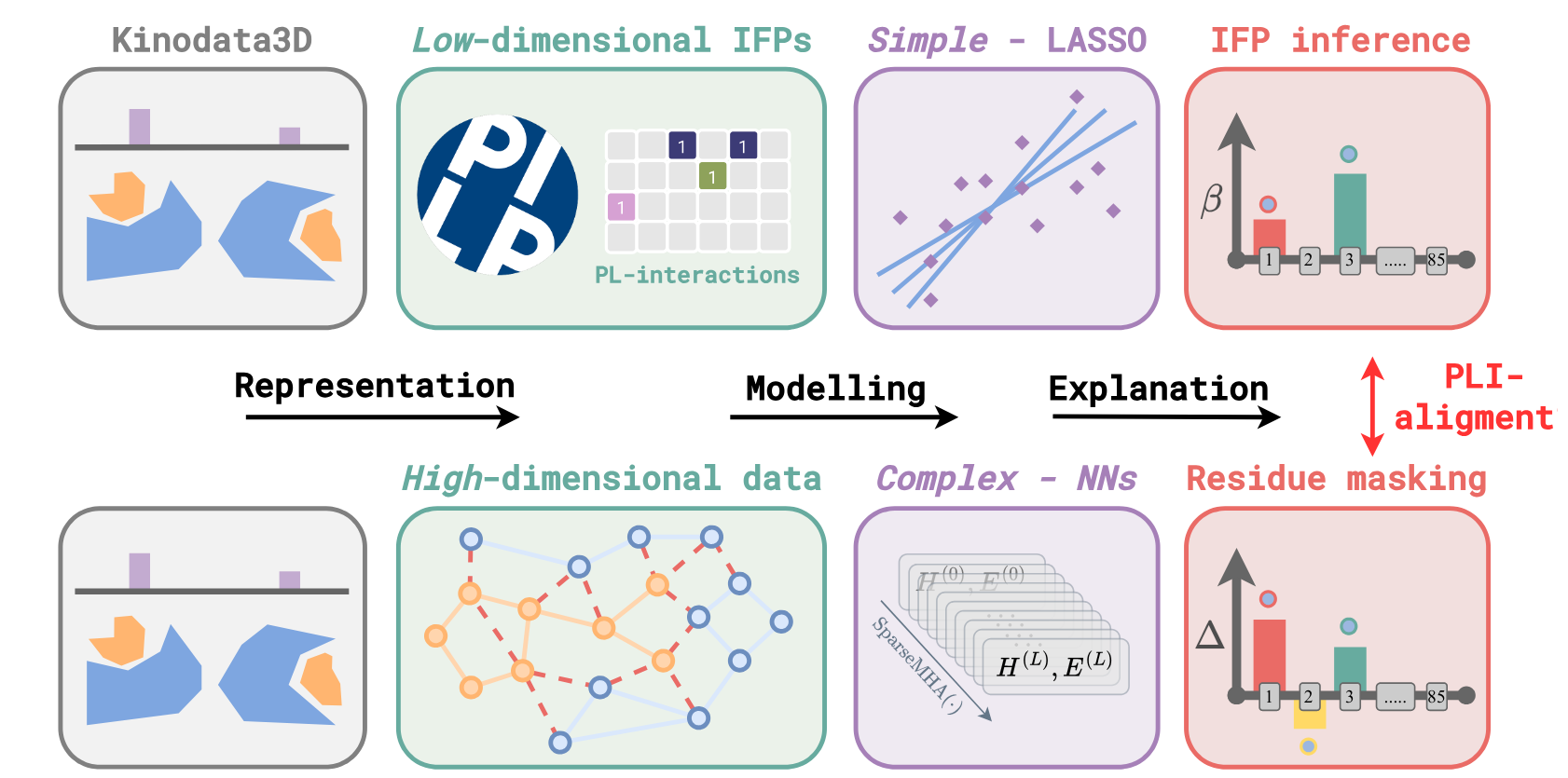
Accurately modeling interactions between small molecules and proteins using machine learning (ML) remains a central challenge in modern drug discovery. In particular, achieving and reliably assessing generalization in ML-based mod- els for binding affinity prediction has proven difficult. Focusing on the protein kinase domain, we investigate both the predictive performance and biophysical plausibility of graph neural networks (GNNs) trained to predict binding affinity, with and without access to docked protein–ligand complexes. To this end, we introduce a model-agnostic explainable AI framework that interprets model pre- dictions by attributing them to specific residues within the ATP-binding pocket of kinases. On the basis of discrete interaction fingerprints (IFPs), we employ robust statistical methods and feature selection to derive reference interaction profiles that serve as ground-truth explanations. Our analysis identifies 131 dis- tinct residues across 29 unique kinases that play key roles in linking IFP and binding affinity data. This enables a complementary benchmark, spanning nearly 10,000 docked complexes, to quantitatively assess model quality not only in terms of predictive accuracy but also biophysical alignment. The benchmark quantifies to what extent the model’s predictive mechanism aligns with meaningful bio- physical mechanisms—specifically, whether it recognizes protein regions actively involved in ligand binding. In a pilot study, we find that models incorporating 3D structural features exhibit moderate biophysical alignment, whereas baseline models without 3D information show no significant alignment, underscoring the structural nature of molecular binding, and importance thereof for training GNN models.